- 上一篇:淡水鱼鱼鳞中嘌呤和嘧啶类化合物同时测定方法研究
- 下一篇:年产1000吨无水亚硫酸钠的工艺设计
摘 要: 采用密度泛函数理论计算一系列三甲基二苯并噻吩化合物的标准电极电势及分子的极性。分子的偶极矩(D)和S原子的Mülliken电荷(MC)之间的关系为:D=5.035 +2.081,相关系数为0.992。大部分甲基化的二苯并噻吩分子极性增大,且标准电位减小。60904
毕业论文关 键 词 : 三甲基二苯并噻吩,标准电位,偶极矩
Abstract :Density functional theory was performed for the calculation of standard electrode potential and molecular polarity for a series of trimethyldibenzothiophenes. The correlation between the dipole moments (D) and the Mülliken charges (MC) of S atom was obtained as D=5.035 +2.081 with the higher correlation coefficients of 0.992. Most methyl groups on the aromatic rings in trimethyldibenzothiophenes enhance the molecular polarity and reduce the standard potentials.
Keywords: trimethyldibenzothiophene, standard electrode potential, dipole moments
1 前言 3
2 计算方法 4
3 结果与讨论 4
3.1分子几何构型对DBTRX反应活性的影响 4
3.2原子电荷和偶极矩之间的关系 5
3.3 计算标准电极电势 8
结 论 11
参考文献 12
致 谢 14
1 前言
加氢脱硫(HDS)可以有效地去除燃油中大部分的含硫化合物,但是很难取出苯并噻吩类硫化物而获得深度脱硫[1-3]。近几年,氧化脱硫(ODS)得到世人的广泛关注, 氧化脱硫技术可以有效地去除石油中芳香烃含硫成分,并且获得极低含硫燃料[4-6]。在氧化脱硫的反应中,通过亲电加成将二价硫氧化形成六价硫,使噻吩类硫化物分子极性增大,从而可以从石油中分离[7, 8]。
在石油中已经发现二苯并噻吩和它的三甲基衍生物 [9-11],根据分子对称性,其同分异构体 (DBTRX, X=1~29)和其原子编号见图1,其氧化态标记为DBTOX。
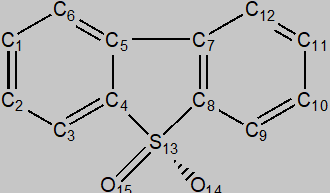
图1 DBTRX的分子结构和原子编号
H2O2在氧化脱硫(ODS)反应中是氧化剂,在氧化DBTRX 中可表示为:
上反应的平衡常数(K)可以由DBTOx/DBTRx 及H2O2/H2O ( 和 )的标准电势和电子转移数(n)来计算:
(25℃, 1atm; =1.77 ).
可见 决定了该反应进行的程度。此外,在H2O2水溶液中DBTRX极性影响反应速率。因此,ODS中DBTOX/DBTRX的标准电位和其极性是至关重要的。
密度泛函理论(DFT)是量子化学中的一个重要方法。氧化还原反应的标准吉布斯自由能变(⊿rGsolu)可以由反应物和产物在水中的吉布斯自由能(Gsolu) 获得,并且对于未知的标准电极电势( )可以由⊿rGsolu、电子转移数n、法拉第常数(F)和参比电极的标准电极电势( )求得:
⊿rGsolu(298.15K,1atm) = -nF( - ).
本文采用密度泛函理论(B3LYP)和6-31+ + g(d,p)基组计算分子的偶极矩和DBTOX /DBTRX的 。
2 计算方法
所有计算均使用采用Gaussian04 软件进行。为了节约计算时间,以密度泛函理论(B3LYP)和6-31+ + g(d,p)基组及极性连续模型(PCM) 在水中优化DBTOX、 DBTRX、 对苯二醌(BQ)、对苯二酚(HBQ)和 H2O分子的几何构型,所有优化计算无几何限制,并通过振动频率分析进一步确认分子结构的合理性。
3 结果与讨论
-
1,7-二(N-(吡啶-2-甲基))胺甲...
-
三组分反应合成1,7-菲罗啉衍生物
-
三组分一锅法合成多取代...
-
三氟苯与取代苯胺的亲核反应研究
-
咪唑并邻菲罗啉-亚铜-1,...
-
三组分反应合成呋喃并咪唑并喹啉衍生物
-
N-甲基芳胺参与的多取代吡啶衍生物的合成
java+mysql车辆管理系统的设计+源代码
电站锅炉暖风器设计任务书
河岸冲刷和泥沙淤积的监测国内外研究现状
当代大学生慈善意识研究+文献综述
杂拟谷盗体内共生菌沃尔...
乳业同业并购式全产业链...
大众媒体对公共政策制定的影响
酸性水汽提装置总汽提塔设计+CAD图纸
中考体育项目与体育教学合理结合的研究
十二层带中心支撑钢结构...